Importer du Matériel Médical : Conformité et Sécurisation (Guide 2026)


Vous souhaitez importer du matériel médical pour équiper un établissement de santé, développer une activité de distribution ou répondre à un appel d'offres ? C'est une démarche aux enjeux considérables : entre réglementation européenne renforcée, exigences de traçabilité et risques liés au choix du fournisseur, les erreurs peuvent coûter très cher — financièrement, juridiquement, et surtout en termes de sécurité des patients.
Pourtant, avec une préparation rigoureuse et un accompagnement adapté, importer des dispositifs médicaux depuis la Chine, la Turquie ou d'autres zones d'approvisionnement reste une opportunité réelle pour les PME et ETI françaises souhaitant optimiser leurs achats.
Ce guide 2026 vous donne les clés pour comprendre le cadre réglementaire, identifier les points de vigilance et sécuriser chaque étape de votre projet d'importation.
Pourquoi Importer du Matériel Médical ?
Le marché mondial des équipements médicaux représentait plus de 469 milliards de dollars en 2025, avec des projections dépassant 1 000 milliards d'ici 2035. Cette croissance soutenue s'explique par le vieillissement des populations, la montée des maladies chroniques et l'essor des technologies de santé.
Pour les acheteurs professionnels français, l'importation de matériel médical répond à plusieurs logiques :
- Optimisation des coûts d'équipement : les fabricants asiatiques, notamment chinois, proposent des équipements dont les prix de revient peuvent être significativement inférieurs aux équivalents européens, sous réserve que la conformité réglementaire soit vérifiée
- Accès à une gamme élargie : certains équipements spécialisés ne sont disponibles qu'auprès de fournisseurs étrangers
- Délais d'approvisionnement maîtrisés : avec un partenaire local bien sélectionné, les délais peuvent être optimisés
- Diversification des sources : réduire la dépendance à un fournisseur unique est une priorité post-COVID pour de nombreux établissements
⚠️ Important : L'importation de matériel médical n'est pas une démarche banale. Chaque projet nécessite une vérification spécifique au regard de la réglementation en vigueur. Une analyse préalable est indispensable avant toute décision d'achat.
Quels Types de Matériel Médical Peut-on Importer ?
La définition d'un dispositif médical au sens du Règlement (UE) 2017/745 (MDR) est large. Elle couvre tout instrument, appareil, logiciel, implant ou accessoire destiné à des fins médicales — diagnostic, prévention, traitement ou compensation d'un handicap.
Voici les grandes familles de matériel médical importable, selon le type d'équipement :
| Catégorie | Exemples | Niveau de complexité réglementaire |
|---|---|---|
| Mobilier et équipements non invasifs | Lits médicalisés, fauteuils roulants, tables d'examen | Modéré |
| Consommables médicaux | Gants, masques, seringues sans aiguille, électrodes ECG | Modéré |
| Équipements de diagnostic | Tensiomètres, thermomètres, oxymètres, ECG | Élevé |
| Équipements thérapeutiques | Respirateurs, machines de dialyse, couveuses | Très élevé |
| Implants et dispositifs invasifs | Prothèses, cathéters, stimulateurs cardiaques | Très élevé + organisme notifié obligatoire |
| Équipements de protection individuelle médicaux | Blouses, sur-chaussures, protections | Variable |
💡 Règle de base : Plus un dispositif est invasif et plus son usage est critique, plus les exigences réglementaires sont strictes. Selon le type d'équipement, les démarches de conformité varient considérablement.
La Réglementation Applicable : Ce Que Tout Importateur Doit Savoir
Du Régime Directive au Règlement MDR : Un Cadre Renforcé
La directive historique 93/42/CEE relative aux dispositifs médicaux a été remplacée par le Règlement (UE) 2017/745 (dit "MDR"), entré en application le 26 mai 2021. Contrairement à une directive, un règlement européen s'applique directement dans tous les États membres, sans transposition nationale.
Ce changement a des implications majeures pour les importateurs :
- Exigences documentaires renforcées : dossier technique, évaluation clinique, système de surveillance post-commercialisation
- Traçabilité totale via le système EUDAMED et les identifiants UDI
- Responsabilités étendues pour les importateurs et distributeurs (Articles 13 et 14 du MDR)
📊 +469 milliards USD en 2025, vers 1 000 Mds d'ici 2035 - Marché mondial des équipements médicaux
Les Classes de Dispositifs Médicaux : Le Cœur de la Réglementation
Le MDR classe les dispositifs médicaux en 4 classes de risque, définies par l'Annexe VIII du règlement. Cette classification conditionne directement les procédures de conformité applicables :
| Classe | Niveau de risque | Exemples | Organisme notifié requis |
|---|---|---|---|
| Classe I | Faible | Lève-personnes, scalpels, électrodes ECG, gants d'examen | Non (sauf stérile ou avec fonction de mesurage) |
| Classe IIa | Modéré | Tensiomètres, thermomètres, aiguilles, tubes d'anesthésie | Oui |
| Classe IIb | Élevé | Respirateurs, couveuses, oxymètres, machines de dialyse | Oui |
| Classe III | Critique | Prothèses articulaires, cathéters cardiaques, stimulateurs cardiaques | Oui — procédure renforcée |
⚠️ Attention : La classification n'est pas toujours évidente. Un même type d'équipement peut relever de classes différentes selon son usage précis. Une analyse préalable est nécessaire pour déterminer la classe applicable à votre produit spécifique.
Les Échéances Réglementaires 2026-2028 à Connaître
Le calendrier de transition vers le MDR s'étale selon les classes :
- 26 mai 2026 : Dispositifs de classe III implantables sur mesure
- 31 décembre 2027 : Dispositifs de classe III et IIb implantables
- 31 décembre 2028 : Dispositifs de classe IIb (autres), IIa et I
📊 Échéances critiques jusqu'en 2028 selon la classe du dispositif - Transition MDR
La Norme ISO 13485 : Référentiel Qualité Incontournable
La norme ISO 13485:2016 définit les exigences d'un Système de Management de la Qualité (SMQ) spécifique aux dispositifs médicaux. Elle concerne les fabricants, mais aussi les importateurs et distributeurs qui doivent structurer leurs processus qualité.
Pour un importateur, cela implique notamment :
- Des procédures de vérification des produits avant mise sur le marché
- Un registre de traçabilité des dispositifs
- Des processus de gestion des non-conformités et des réclamations
- Une documentation contractuelle avec les fournisseurs
Comment Vérifier la Conformité CE d'un Dispositif Médical Importé ?
La conformité CE d'un dispositif médical n'est pas un simple logo à vérifier visuellement. Pour un importateur, selon l'article 13 du MDR, plusieurs vérifications sont obligatoires avant toute mise sur le marché européen :
Les Vérifications Documentaires Essentielles
1. La Déclaration de Conformité UE
Le fabricant doit fournir une déclaration attestant que le produit répond aux exigences essentielles de sécurité et de performance. Ce document doit mentionner : le nom et l'adresse du fabricant, la description du dispositif, les normes harmonisées utilisées, et l'identité du signataire.
2. Le Certificat de l'Organisme Notifié (pour les classes IIa, IIb, III)
Pour les dispositifs de classe supérieure à I, un organisme notifié accrédité doit avoir évalué et certifié la conformité. Vérifiez :
- La validité et la portée exacte du certificat
- L'identité de l'organisme notifié (consultez la base NANDO de la Commission européenne)
- La correspondance entre le certificat et le produit exact que vous importez
3. L'Enregistrement EUDAMED
La base de données européenne EUDAMED permet de vérifier l'enregistrement des dispositifs et des acteurs économiques. En tant qu'importateur, vous avez l'obligation de vous y enregistrer.
4. L'Étiquetage et la Notice
L'étiquetage doit être en français (ou dans la langue du pays de destination) et contenir toutes les informations réglementaires requises.
🔗 Pour aller plus loin sur ce sujet, consultez notre article dédié : Guide complet 2026 : Comment vérifier la validité d'un certificat CE pour un produit importé ?
Sécuriser le Choix de Votre Fournisseur d'Équipements Médicaux
Le choix du fournisseur est l'étape la plus critique d'un projet d'importation de matériel médical. Un fournisseur non qualifié peut vous exposer à des risques majeurs : produits non conformes, refus en douane, responsabilité juridique, voire mise en danger des utilisateurs.
Les Critères de Qualification d'un Fournisseur Médical
| Critère | Ce qu'il faut vérifier | Niveau de priorité |
|---|---|---|
| Certification ISO 13485 | Certificat valide, portée couvrant le produit concerné | 🔴 Indispensable |
| Enregistrement NMPA (Chine) | Licence de fabrication de dispositifs médicaux | 🔴 Indispensable |
| Dossier technique disponible | Documentation technique complète transmissible | 🔴 Indispensable |
| Références clients UE | Expérience d'exportation vers l'Europe documentée | 🟠 Très important |
| Capacité d'audit | Acceptation d'audits qualité sur site | 🟠 Très important |
| Solidité financière | Vérification via rapports ou agences spécialisées | 🟡 Important |
Les Signaux d'Alerte à Ne Pas Ignorer
- Fournisseur incapable de fournir la documentation technique complète
- Certificats CE "génériques" ne correspondant pas précisément au produit
- Refus ou impossibilité d'audit sur site
- Prix anormalement bas par rapport au marché (souvent signe de compromis sur la qualité)
- Absence d'expérience documentée à l'export vers l'UE
🔗 Découvrez notre approche du sourcing fournisseur international et comment nous sécurisons la qualification de vos partenaires.
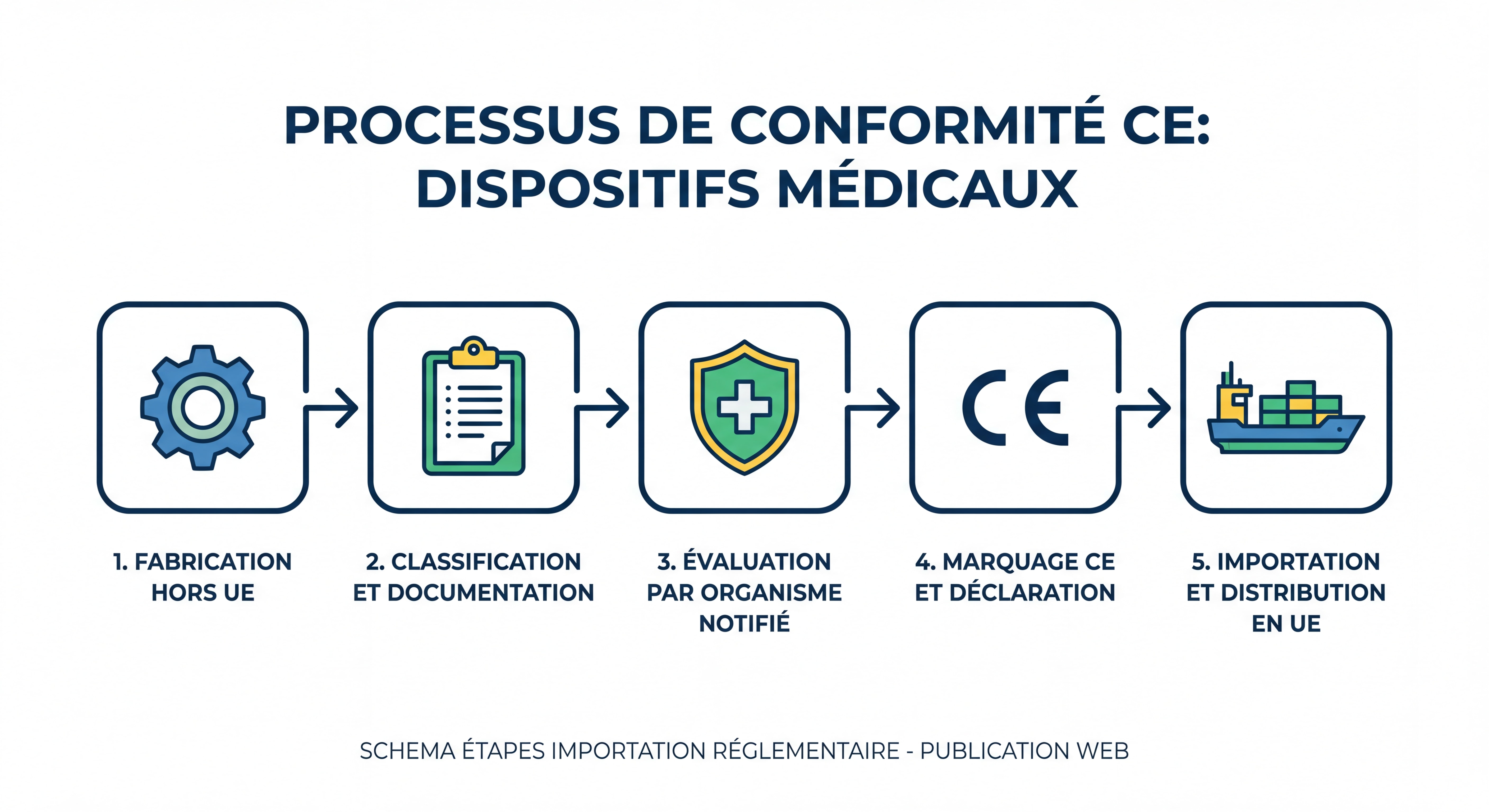
Le Processus d'Importation Étape par Étape
Importer du matériel médical en France nécessite de suivre un processus structuré. Voici les grandes étapes, étant entendu que chaque projet nécessite une vérification spécifique selon la nature du dispositif et son origine géographique :
Étape 1 — Qualification réglementaire du produit
Déterminer la classe du dispositif médical, identifier le règlement applicable (MDR ou IVDR), et lister les exigences de conformité spécifiques.
Étape 2 — Sourcing et qualification du fournisseur
Identifier des fournisseurs potentiels, vérifier leurs certifications, réaliser un audit qualité (sur site ou documentaire), et obtenir l'ensemble de la documentation technique.
Étape 3 — Vérification de la conformité CE
Analyser la déclaration de conformité, vérifier le certificat de l'organisme notifié, contrôler l'étiquetage et la notice en langue française.
Étape 4 — Enregistrement EUDAMED
S'enregistrer en tant qu'importateur dans la base de données EUDAMED et vérifier l'enregistrement du fabricant et du dispositif.
Étape 5 — Formalités douanières et logistique
Classement tarifaire (codes NC), droits de douane applicables, TVA à l'importation (autoliquidation depuis 2022), et organisation du transport dans le respect des conditions de stockage requises.
Étape 6 — Mise en place du SMQ importateur
Structurer les procédures internes : traçabilité, gestion des non-conformités, surveillance post-commercialisation, et contrats avec le fabricant.
Étape 7 — Mise sur le marché et suivi
Apposer les informations d'importateur sur l'étiquetage si nécessaire, maintenir les registres réglementaires, et assurer la vigilance continue.
📊 Articles 13 et 14 du Règlement UE 2017/745 — vérification, traçabilité, enregistrement EUDAMED - Obligations importateur MDR
Points de Vigilance Essentiels pour l'Importateur
Au-delà du processus standard, certains points méritent une attention particulière :
⚠️ La Responsabilité Étendue de l'Importateur
Sous le MDR, si vous apposez votre marque sur un dispositif importé sans accord explicite avec le fabricant, vous devenez fabricant au sens réglementaire et supportez l'intégralité des obligations correspondantes. Cette situation, fréquente dans les pratiques de "marque blanche", doit être analysée avec soin.
⚠️ La Cybersécurité des Dispositifs Connectés
Depuis 2026, les dispositifs médicaux connectés sont soumis à des exigences croissantes en matière de cybersécurité (NIS2, et à partir de décembre 2027, le Cyber Resilience Act). Si vous importez des équipements communicants, une analyse préalable de leur sécurité informatique est nécessaire.
⚠️ Les Conditions de Transport et de Stockage
Certains équipements médicaux nécessitent des conditions spécifiques de transport (température contrôlée, protection contre les chocs). Une rupture de la chaîne de conditions peut invalider la conformité du produit.
⚠️ La Vigilance Post-Commercialisation
En tant qu'importateur, vous avez l'obligation de signaler à l'ANSM tout incident grave ou problème de sécurité constaté sur les dispositifs que vous avez mis sur le marché.
📞 Vous avez un projet d'importation de matériel médical ?
Avant de vous lancer, échangeons sur votre besoin spécifique. Synapse Global Trade vous accompagne dans l'analyse de faisabilité et la sécurisation de votre démarche.
→ Échanger sur votre besoin
Questions Fréquentes (FAQ)
Peut-on importer du matériel médical depuis la Chine pour le vendre en France ?
Oui, c'est possible, mais cela nécessite une analyse préalable rigoureuse. Le matériel médical importé depuis la Chine doit être conforme au Règlement européen MDR (UE 2017/745) et porter le marquage CE valide. Le fournisseur chinois doit disposer d'une certification ISO 13485 et d'une documentation technique complète. En tant qu'importateur basé dans l'UE, vous avez des obligations spécifiques de vérification, d'enregistrement EUDAMED et de traçabilité. Chaque projet nécessite une vérification spécifique selon la classe du dispositif concerné.
Quelle est la différence entre la directive 93/42/CEE et le règlement MDR 2017/745 ?
La directive 93/42/CEE était l'ancien cadre réglementaire applicable aux dispositifs médicaux en Europe. Elle a été remplacée par le Règlement (UE) 2017/745 (MDR), entré en application le 26 mai 2021. La différence fondamentale est juridique : un règlement s'applique directement dans tous les États membres sans transposition nationale. Sur le fond, le MDR renforce considérablement les exigences : évaluation clinique obligatoire, surveillance post-commercialisation systématique, traçabilité via EUDAMED/UDI, et responsabilités étendues pour tous les opérateurs économiques, y compris les importateurs.
Un importateur doit-il être certifié ISO 13485 ?
La certification ISO 13485 n'est pas formellement obligatoire pour un importateur, mais le MDR (articles 13 et 14) impose la mise en place d'un système de gestion de la qualité. L'ISO 13485 est le référentiel recommandé pour structurer ce SMQ. En pratique, de nombreux clients professionnels et établissements de santé exigent cette certification de leurs fournisseurs. Une analyse préalable de votre situation et de vos marchés cibles est nécessaire pour déterminer le niveau de formalisation adapté.
Quels sont les risques en cas d'importation de matériel médical non conforme ?
Les risques sont multiples et sérieux : retrait du marché et destruction des produits, amendes et poursuites pénales pour mise en danger d'autrui, responsabilité civile en cas d'incident, interdiction d'exercer l'activité d'importateur, et bien sûr, atteinte à la réputation de votre entreprise. L'ANSM dispose de pouvoirs de contrôle et de sanction étendus. C'est pourquoi une vérification documentaire complète avant toute importation est indispensable.
Comment Synapse Global Trade peut-il m'aider à importer du matériel médical ?
Synapse Global Trade accompagne les PME et ETI françaises dans leurs projets d'importation depuis la Chine, l'Europe, la Turquie et l'Asie. Notre intervention couvre l'analyse de faisabilité réglementaire, la qualification et l'audit des fournisseurs, la vérification documentaire de conformité, la gestion des formalités douanières et logistiques, et le suivi qualité. Chaque mission est adaptée à votre projet spécifique — nous ne promettons jamais de résultats sans avoir réalisé une analyse préalable de votre dossier.
Chiffres Clés
📊 +469 milliards USD : taille du marché mondial des équipements médicaux et de diagnostic en 2025, avec une projection dépassant 1 000 milliards USD d'ici 2035 (Source : Research Nester, 2026)
💡 4 classes de risque (I, IIa, IIb, III) définissent les exigences de conformité applicables à chaque dispositif médical selon le Règlement MDR (UE) 2017/745
⚖️ 2026-2028 : période de transition critique pour la mise en conformité complète au MDR — les échéances varient selon la classe du dispositif (Source : Intertek / EUR-Lex, 2026)
🔒 Articles 13 et 14 du MDR : obligations spécifiques des importateurs — vérification, enregistrement EUDAMED, traçabilité et vigilance post-commercialisation (Source : ANSM, 2026)
Conclusion : Une Opportunité Réelle, Sous Réserve d'une Préparation Rigoureuse
Importer du matériel médical représente une opportunité stratégique pour les PME et ETI françaises souhaitant optimiser leurs coûts d'équipement ou développer une activité de distribution dans le secteur de la santé. Mais c'est aussi un domaine où l'improvisation peut avoir des conséquences graves.
Les points essentiels à retenir :
- ✅ Le cadre réglementaire est désormais celui du Règlement MDR (UE) 2017/745, plus exigeant que l'ancienne directive 93/42/CEE
- ✅ La classification du dispositif (I, IIa, IIb, III) détermine les procédures de conformité applicables — une analyse préalable est toujours nécessaire
- ✅ En tant qu'importateur, vous avez des obligations propres de vérification, d'enregistrement et de traçabilité
- ✅ La qualification du fournisseur est l'étape la plus critique : exigez la documentation complète et n'acceptez jamais un certificat CE sans le vérifier
- ✅ Chaque projet est unique : selon le type d'équipement, le pays d'origine et votre positionnement dans la chaîne de distribution, les démarches varient
Synapse Global Trade accompagne les PME et ETI françaises dans leurs projets d'importation internationale avec une approche structurée, prudente et orientée résultats. Nous intervenons en amont pour sécuriser vos décisions — pas après les erreurs.
📋 Vous avez un projet d'importation de matériel médical ?
Partagez-nous votre besoin et bénéficiez d'une analyse de votre projet par nos experts.
→ Demander une analyse de projet